Management of Euglycemic Diabetic Ketoacidosis - Insulin is the Secret Sauce
This post will review the presentation, workup, and treatment of euglycemic diabetic ketoacidosis (eDKA). Particular attention is spent discussing refractory acidosis, treatment failure, and insulin dosing.
TL;DR
- eDKA is a medical emergency and the management is NOT straightforward
- SGLT-2 inhibitor use and pregnancy are major risk factors for eDKA
- Patients may NOT present with ketonuria if they are on an SGLT-2 inhibitor
- Treating eDKA demands balancing the risks of hypoglycemia and insulin deficiency
- Refractory anion gap metabolic acidosis should be a HUGE RED FLAG that the insulin dose is NOT HIGH ENOUGH - you MUST give more dextrose AND more insulin!
- The most common pitfall in the management of eDKA is NOT GIVING ENOUGH INSULIN. Start with at least 0.05 units/kg/hour along with IV dextrose.
- MAKE SURE TO GIVE AT LEAST 5 GRAMS/HOUR OF DEXTROSE - this will be 100 mL per hour of a 5% dextrose solution - most patients need 5-10 g/h.
- D5 = 50 g/L; D10 = 100 g/L; D20 = 200g/L ... and so on
Intro
Euglycemic diabetic ketoacidosis (eDKA) is defined as the presence of an anion gap metabolic acidosis, ketosis, a blood sugar under 250 mg/dL, and no other identified etiology.
eDKA is caused by an imbalance of insulin and glucagon that disrupts the steady state of ketones in the body. The risk factors and treatment are a direct result of this fact.
I love physiology, but feel free to skip this next part if it's not your thing.
Insulin reduces ketone production
Ketones are made in the liver and used in peripheral tissue. They are made in the mitochondria from Acetyl-CoA. Fat breakdown in tissues and oxidation in the liver leads to huge amounts of Acetyl-CoA in the liver cytosol. The acetyl-CoA is transported into the mitochondria where ketone formation happens.
Insulin decreases fat breakdown and inhibits acetyl-CoA transport into the mitochondria. Therefore insulin REDUCES ketone production.
Glucagon increases ketone production
Glucagon causes increased fat breakdown and stimulates ketone production in the liver directly!
SGLT-2 inhibitors reduce insulin AND increase glucagon
Insulin is secreted by beta cells in the pancreas. The strongest signal for insulin production is HIGH BLOOD SUGAR.
SGLT-2 inhibitors work at the proximal tubule of the kidney. They inhibit the pump that reabsorbs glucose and excretes sodium. In this way they directly reduce the glucose in the blood. Because there is less glucose, there is less insulin.
Pre-hepatic insulin-glucagon ratio is reduced by up to 25% in patients on SGLT-2 inhibitors.
More glucose reaches the distal renal tubule when a patient is on a SGLT-2 inhibitor. These distal transporters consequently absorb more glucose than usual. As glucose is absorbed, sodium is exchanged into the renal tubules. This effect causes a net positive charge in the tubules. This electrical gradient results in MORE ketone (+) absorption in the distal renal tubule.
Distal renal tubular absorption of ketones can cause reduced or ABSENT ketonuria in patients with eDKA on SGLT-2 inhibitors!
There are some data that suggest SGLT-2 inhibitors may directly increase glucagon production in the alpha cells of the pancreas. These are animal studies only, so presence of this effect is not absolute.
The final effect of SGLT-2 inhibitors on ketone promotion is from osmotic diuresis. Hypovolemia increases beta-1 agonist activity which reduces insulin and increases glucagon - thus propagating the problem.
In summary, SGLT-2 inhibitors cause a GLUCOSE deficit and shift the balance of insulin and glucagon towards ketosis. They cause renal reabsorption of ketones, osmotic diuresis, and may directly stimulate glucagon production. As a result of these effects, patients on these drugs have an increased likelihood of euglycemic DKA.
Pregnancy reduces glucose and promotes ketosis
DKA in pregnancy is a medical and obstetric emergency with up to a 35% risk of serious complications including maternal or fetal demise. High clinical suspicion and early identification are the best ways to take care of these patients.
Plasma volume increases 40% by the end of pregnancy and GFR increases 60%. This causes glucose dilation and more renal glucose filtration. In addition, placental glucose uptake increases dramatically through pregnancy. Caloric needs by the third trimester can increase by up to 300 kilocalories per day!
Less glucose = less insulin
Less insulin = more ketones
The placenta also secretes human placental lactogen, placental insulinase, and progesterone. All of these hormones increase insulin resistance and promote fat breakdown and hepatic oxidation.
As a result of these effects, 30% of pregnant patients with DKA are euglycemic on presentation!
Presentation and Workup
eDKA presents similar to hyperglycemic DKA. Patients present with GI symptoms of nausea, abdominal pain, vomiting and eventually develop respiratory symptoms and altered mental status.
The bulk of your efforts evaluating a patient with potential eDKA will be evaluating for alternative causes of anion gap metabolic acidosis. A full review of this differential diagnosis is out of the scope of this post. Briefly, you should consider all of the following:
- Lactic acidosis
- Ethanol
- Methanol
- Ethylene glycol
- Salicyate ingestion
- TCA overdose
- Paracetamol induced D-lactic acidosis
- Starvation ketoacidosis
- Alcoholic ketoacidosis
A typical patient with euglycemic DKA will present with a high anion gap metabolic acidosis, ketonuria, and no other cause for their anion gap. In particular, ethanol levels, lactate, and salicylate levels should all be normal! Multiple studies have investigated lactate levels for allcomers with DKA and 95% of the time it will be measured less than 2 mmol/L.
Keep in mind that a urinalysis uses nitroprusside to measure acetone and acetaldehyde, but the main ketone in DKA is beta hydroxybutyrate. A BHB over 3 mmol/L is extremely concerning for a ketoacidosis contributing to the patient's presentation.
In addition to pregnancy and SGLT-2 inhibitor use, additional risk factors may include the following:
- Surgical procedure
- Infection
- Ischemia (stroke, myocardial infarction, ischemic bowel)
- Low carbohydrate intake
- Recent reduction in insulin regimen (?maybe on initiation of an SGLT-2i)
- Underweight with LOW glycogen stores
- Have T1DM or T2DM with severe insulin deficiency
(15% of patients will have NO history of diabetes at presentation)
All of these risk factors compound on each other. This is the reason that eDKA occurs despite many physiologic protective mechanisms.
Treatment
The fundamentals of treatment for eDKA are similar to those of DKA. The main difference is that you MUST give insulin despite low blood sugars and supplement with dextrose.
Monitoring
Patients with eDKA or DKA die from electrolyte abnormalities. So, the first thing you need to order for a sick patient is hourly blood sugars and every 2 hour venous blood gas, basic metabolic panel, magnesium, phosphorus, and ionized calcium. The large majority of patients with euglycemic DKA should be admitted to an ICU and otherwise definitely deserve a telemetry bed.
Give fluids
Unless there is a strong contraindication, give the patient a robust fluid bolus. Hypovolemia propagates ketosis through beta-1 mediated insulin-glucagon imbalance.
Patients who are hyperglycemic often also have hypochloremia, so saline is appropriate for them. In contrast, euglycemic patients are more likely to have NORMAL chloride. So, a chloride rich bolus may contribute to a concurrent non-anion gap metabolic acidosis.
Favor administration of a balanced crystalloid fluid bolus
Electrolytes
Aggressively trend and replete potassium, magnesium, and phosphorus as you would with a typical case of DKA.
If potassium is ever less than 3.5 mmol/L, then stop any source of insulin and don't resume until the potassium is better.
Give Insulin and Dextrose
Insulin is the cure for the ketoacidosis component of DKA. You must shift the insulin-glucagon balance to favor ketolysis. The change in anion gap is linearly correlated with the insulin dose (figure 1).
Low dose insulin administration is insufficient to stop ketone formation! Ketosis DOES NOT RESOLVE until ketone clearance exceeds production.
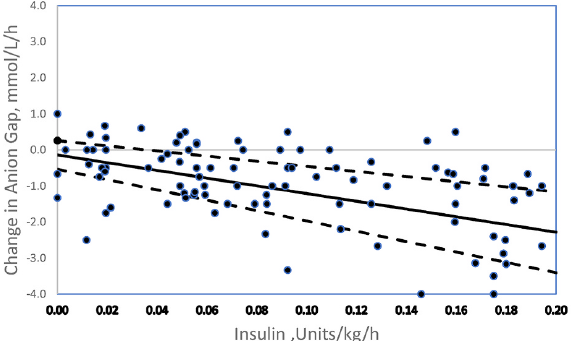
There are no society guidelines that comment on insulin dosing for euglycemic DKA so management is largely extrapolated from management of hyperglycemic DKA.
Most standard DKA protocols reduce insulin dose as blood sugars go down. Consequently, using a standard DKA protocol is likely to UNDERDOSE the insulin.
Start insulin at 0.05 - 0.1 units/kg/hour
I would recommend starting the insulin at a conservative dose of 0.05 units/kg/hour. This provides a good balance between the risks of hypoglycemia and insufficient insulin.
Start a 5% dextrose containing solution at 100-200 mL/hour
Dextrose needs are determined by the amount of insulin needed to sufficiently inhibit ketone production. Estimates are based on retrospective data and one study that measured radioactive glucose uptake in patients with DKA on insulin.
Patients with DKA should get about 5-10 g/h of dextrose. Since D5 = 50 g/L; D10 = 100 g/L; D20 = 200g/L ... and so on, that means patients should be on the equivalent of 100-200 mL/hour of D5.
If the patient has contraindication to receiving large volumes of dextrose containing fluids, such as hyponatremia, then higher concentrations and lower volumes would be better.
Cases from literature
Alalgy et al, 2023
A 26 year old woman at 28 weeks of her pregnancy and a medical history of type 1 diabetes for 5 years on insulin and metformin presented with one day of abdominal discomfort, nausea, and multiple episodes of vomiting in the setting of insulin non-compliance.
Initial vitals were temp 97F, HR 105, RR 26, BP 128/78. On exam she appeared dehydrated. Her abdomen was soft and non-tender throughout.
She was empirically treated with 3L of LR and had labs collected which later showed pH 7.17, PCO2 20, Bicarbonate 7.4, lactate 1.7, glucose 190 mg/dL, and potassium of 4.4 mg/dL. Her urinalysis showed 4+ ketones.
She denied any carbohydrate restriction, alcohol/toxic alcohol use, or tylenol/salicylate use. No infectious causes were identified.
A diagnosis of eDKA was made and the patient was concurrently started on insulin at 0.05 units/kg/h and a D5 dextrose solution. The insulin infusion was later increased to 0.1 units/kg/h with an intent to hasten acidosis recovery in the setting of her pregnancy.
Azman et al, 2022
A 60 year old man with past medical history of non-insulin dependent type 2 diabetes, hypertension, and hyperlipidemia presented to the emergency department with chest pain. He was ultimately diagnosed with an NSTEMI and treated with DAPT. Empagliflozin was started given history of diabetes.
He presented again the hospital six days later with sudden onset headache and cranial nerve deficits. An MRI demonstrated a pituitary macroadenoma and evidence of pituitary apoplexy. The patient was started on treatment for hypopituitarism which included hydrocortisone.
The following day the patient became hypotensive and tachypneic. He was started on norepinephrine and non-invasive ventilation and moved to the ICU.
His next set of labs demonstrated an anion gap of 26, potassium 4.5, pH 7.059, and bicarbonate of 6.6. Lactate level was 1.0. Furthermore, blood cultures grew gram positive cocci. His urinalysis was positive for 4+ ketones.
A diagnosis of euglycemic DKA was suspected. Empagliflozin was held and the patient was started on an insulin infusion.
Over the next 10 hours the patient had refractory acidosis. He received extensive fluid resuscitation and the team planned to initiate CRRT. At this time he was started on a 10% dextrose infusion and insulin infusion was increased to 5 units/h.
Within 4 hours of initiating these treatments the patient made a drastic recovery with bicarb increasing to 14.1 and pH to 7.3.
Jaber et al, 2019
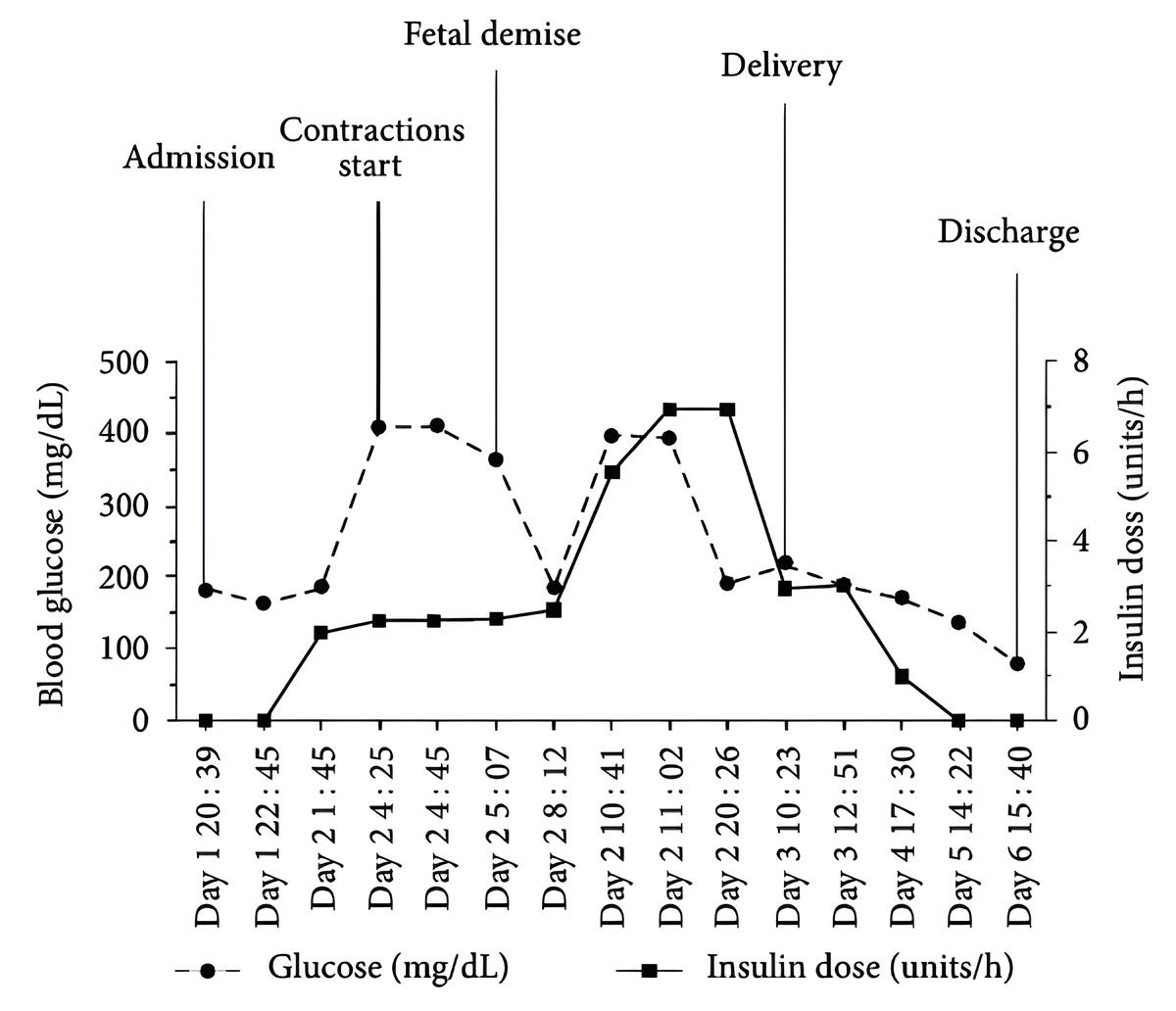
Trigger warning of fetal demise for this case
A 30 year old G2P1 at 32 weeks and 3 days of gestation and a medical history notable for type 1 diabetes on an insulin pump and a prior episode of DKA. She presented with two days of nausea and vomiting.
On exam she appeared tachypneic and uncomfortable. Her abdomen was soft and non-tender. Fetal heart monitor showed absent variability and recurrent late decelerations.
Her initial lab workup revealed blood sugar of 183 mg/dL, anion gap of 23, pH 7.1, beta hydroxybutyric acid 9.6 mmol/L, and lactate of 0.65 mmol/L.
The patient was admitted and placed on an insulin infusion at 2 units/h and D5 1/2 NS at 250 mL/h based on the facilities DKA protocol.
Over the subsequent 4 hours the patient deteriorated clinically and became more acidotic with a pH of 6.97. Fetal heart tracings deteriorated and the patient was given multiple amps of bicarb. Her insulin drip was increased and her IV fluids were changed to 10% dextrose at 250 mL/h.
Unfortunately, the patient continued to decline and was intubated. At eight hours after admission the fetal heart rate was not able to be detected and fetal demise was declared. At that point the patient's insulin was at 7 units/h. The patient achieved stability for delivery, and after this her insulin requires and acidosis rapidly resolved.
Summary
Euglycemic DKA is caused by an imbalance of insulin and glucagon causing ketone accumulation. In addition to typical risk factors for DKA, those for eDKA include SGLT-2 inhibitor use and pregnancy.
Diagnosis requires ruling out other causes of anion gap metabolic acidosis.
In addition to carefully managing fluid and electrolytes, the treatment of eDKA is focused on giving enough insulin to reverse ketone accumulation and prevent iatrogenic hypoglycemia. If the patient's acidosis is not improving, then they need MORE INSULIN and MORE DEXTROSE.